FDA Orange Book
The FDA Orange Book, formally "Approved Drug Products with Therapeutic Equivalence Evaluations", is the authoritative reference for approved drug products, therapeutic equivalents, and the patent and exclusivity records that govern generic entry. Published since 1980, it is the primary source for understanding when brand-name drugs face generic competition.
Get notified when we publish new research.

The RxDataLab FDA Exclusivity Dataset combines Orange Book records with USPTO patent data, drug label indication text, and monthly change history. Available as a one-time export or annual data license.
Approved Drug Products with Therapeutic Equivalence Evaluations, more commonly known as the Orange Book, is a publication containing FDA approved drugs, associated patent and exclusivity information, and therapeutic equivalents1. Today, the Orange Book is an important reference for pharmaceutical analysts, business development teams, financial analysts, and generic manufacturers tracking when branded drugs face competition. Here we cover the history, contents, and practical use of this publication.
Where did the Orange Book Come From? #
The original Orange Book was created via federal rule making in 19802. In the initial proposal, the FDA cited the following as primary motivations for establishing the publication:
- Education
- Cooperation with state medical authorities
- Combating inflation
Education and State Cooperation
Before the 1970s, when a physician prescribed a brand name drug, most state laws required the pharmacist to dispense exactly that brand of drug, even if a cheaper and equivalent generic option existed. This was a popular state of affairs if you ran a drug company. Indeed, the initial Orange Book proposal described efforts by drug companies and medical societies to confuse the public about therapeutic equivalents, with claims that “special coatings” or “fillers” caused proprietary drugs to be more effective than generics. One comment from a campaign even stated that “chemically equivalent drugs may not have the same effect”, and gave the example of diamonds and coal both being pure carbon3.
Despite these campaigns, from 1970 to 1977, physicians were prescribing drugs based on generic name (the non-brand name of active ingredients) at an increasingly frequent rate, and partly in response to high drug costs, education, and inflation, state laws were changing to allow pharmacists to dispense the lower cost equivalent option4 when available.
By 1979, 40 states and the District of Columbia had enacted so called “drug price substitution laws” — a dramatic turn from virtually all states having “anti-substitution laws” before56. With these new laws and prescribing trends amongst physicians, pharmacists had a greater responsibility to identify (and keep stocked) the appropriate drugs to dispense to patients. But with no authoritative list of approved brand name drugs and therapeutic equivalents, this was a challenging task.
State medical authorities wanted an authoritative list to help providers accurately substitute drugs, and some states, including New York, created their own7. FDA notes that by 1979, 19 states and the District of Columbia had approached FDA to help create state lists — further emphasizing the need for a central, authoritative list.
Inflation
By 1974, inflation had reached nearly 12%, in 1980, it would peak at 13.5%8. At a press conference in 1978, President Carter noted:
One of the very discouraging aspects of our present health care system is the enormous increase in costs that have burdened down the American people. The average increases in cost of health care per year has been more than twice as much as the overall inflation rate.
Encouraging the use of therapeutically equivalent generics represented an opportunity to dramatically cut healthcare costs without sacrificing the quality of care for Americans. And a simple step towards this goal would be letting people know cheaper alternatives exist.
The First Publication
In response to requests from state health officials and medical professionals, the FDA established “Approved Drug Products with Therapeutic Equivalence Evaluations” via federal rulemaking and the first issue was published on October 31, 1980, hence the name529.
The publication was codified with the passage of the Drug Price Competition and Patent Term Restoration Act of 19841011, and the patent information and exclusivity information was also added.

What Type of Information is in the Book? #
The Orange Book and associated electronic resources are currently maintained by CDER.
While the initial Orange Book contained only approved drugs and equivalents, the current Orange Book contains11:
- Approved drug products and therapeutic equivalents
- Patent and exclusivity information for approved drug products
- Approved over the counter drug products
- Drug products approved by CDER (under section 505) but administered by CBER
- Cumulative list of approved products
Importantly, the Orange Book contains only approved drug products regulated by CDER, so tentative approvals and biologics regulated by CBER are not included12. Biologics — including monoclonal antibodies, vaccines, and biosimilars — are tracked separately in the FDA Purple Book, which covers biosimilar and interchangeable product designations for biologics regulated under the Public Health Service Act.
The annual Orange Book publication is available from FDA. The text version is dense and difficult to analyze. Here is figure 2.2 from the 44th edition (2024) as an example:
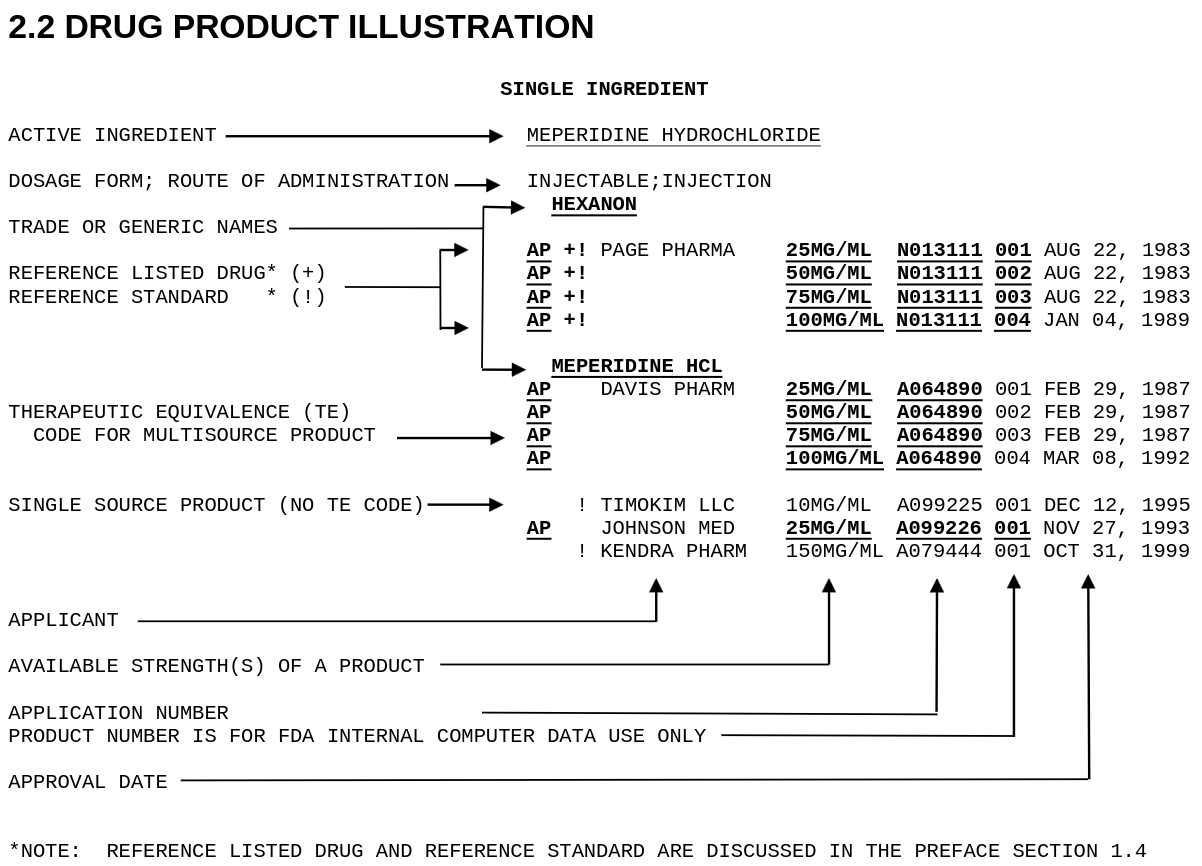
Therapeutic Equivalence (TE) codes are the key to determining whether a product can be substituted for a brand name drug. Codes beginning with “A” indicate equivalence, and codes beginning with “B” indicate the generic is not considered equivalent. The most common TE code is AB — a drug that has met the FDA’s standards for bioequivalence and can be freely substituted. An AA rating indicates no known or suspected bioequivalence problems with conventional dosage forms.
Patent and Exclusivity Information
The Orange Book also contains patent and exclusivity information about prescription and over the counter drug products.
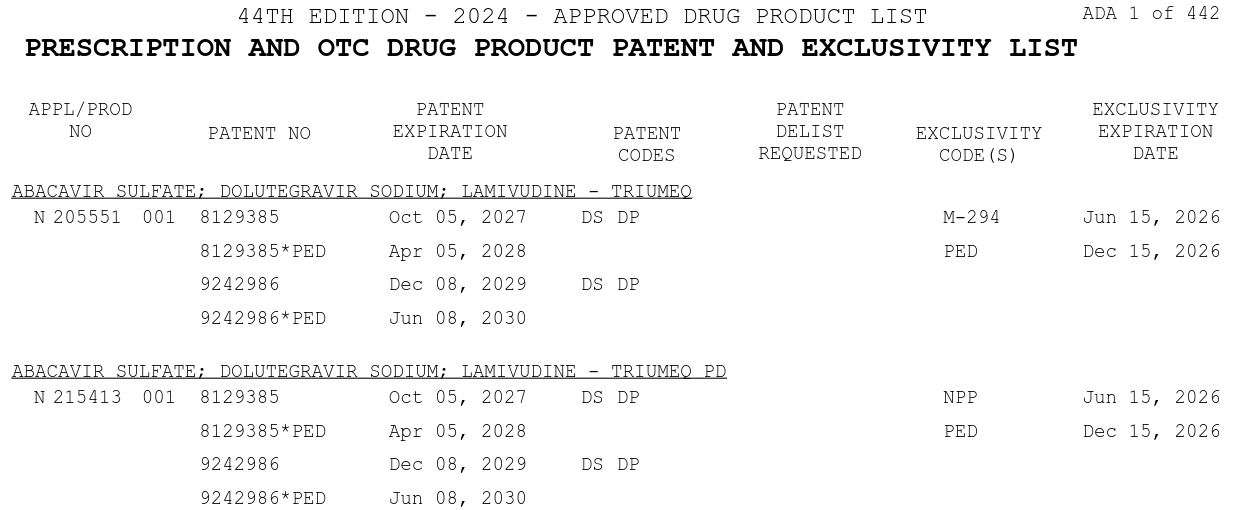
The patent information is provided by NDA applicants and is not independently validated by USPTO or FDA — the agency considers its role purely ministerial. This means NDA sponsors self-report which patents cover their drugs, and listings may be incorrect, outdated, or actively disputed in litigation. Patents within the Orange Book can be, and frequently are, challenged for accuracy13.
Separate from patents, FDA also grants statutory exclusivity — a set of market protections under the FD&C Act that operate independently of patent status. A drug may retain regulatory exclusivity even after its listed patents have expired, and conversely, patents may be invalidated through litigation while exclusivity remains intact. Understanding both is essential for accurately estimating when generic entry can occur.
Reading Patent Expiration and Loss of Exclusivity Data #
The Orange Book is the primary source for estimating when a brand-name drug will face generic competition — but reading it correctly requires understanding what the data represents and where it falls short.
What the expiration dates mean
Each patent listed in the Orange Book carries an expiration date. These are the dates reported by the NDA holder to FDA; they are not independently verified. A listed patent may cover the drug substance, drug product formulation, or a specific method of use, and the same patent can be listed across multiple applications.
Importantly, the last patent expiration date is not necessarily the loss-of-exclusivity (LOE) date. Several factors can move the effective generic entry date earlier or later:
- Regulatory exclusivity extends beyond patents. A new chemical entity receives 5 years of NCE exclusivity from approval, an orphan drug 7 years, and qualifying drugs can receive pediatric extensions of 6 months on top of existing protections. The effective LOE is the later of the last patent expiry or the last exclusivity expiry.
- Paragraph IV patent challenges can accelerate entry. A generic manufacturer can challenge listed patents as invalid or non-infringed under the Hatch-Waxman Act. If successful, the generic may launch before the stated patent expiry. The first successful challenger receives 180 days of marketing exclusivity.
- Patent term extensions can push dates out. The FDA review period may be partially restored via Patent Term Extension (PTE) under 35 U.S.C. § 156, extending patent coverage beyond the original expiry.
- Inter partes review (IPR) proceedings at the USPTO can invalidate patent claims after listing, potentially eliminating protection that the Orange Book still shows as active.
For analytical purposes, a reported LOE estimate can be computed as:
Reported LOE = MAX(latest patent expiry + any pediatric extension, latest exclusivity expiration)
This is how the loe_estimates table in the RxDataLab FDA Exclusivity Dataset works. It is a useful starting point for modeling generic entry windows, but it is not a legal determination — litigation, Paragraph IV outcomes, and authorized generics can all cause actual market entry to differ from the reported date.
Orange Book codes database
The Electronic Orange Book data files expose the underlying records as structured downloads. The FDA publishes separate files for products, patents, exclusivity records, and use codes. The use codes — a two-letter system identifying what aspect of a drug a patent covers — are important for interpreting patent challenges. A patent with use code U-443 covers a specific method of use, while a DS code covers the drug substance itself. Use code patents are frequently the target of “skinny label” strategies where a generic can be approved for non-patented indications while avoiding the method-of-use patent.
The FDA provides plain-English definitions for all active use codes. For structured analysis, the RxDataLab exclusivity dataset includes a patent_use_codes reference table with all current definitions, and the patents table includes the use code and pediatric exclusivity flag for every Orange Book listing.
How can you use the Orange Book? #
The Orange Book was initially intended as a resource to provide medical professionals with a list of approved drugs and their corresponding therapeutic equivalents. In practice, it is now more frequently used to understand the exclusivity and intellectual property restrictions surrounding a particular drug — and specifically, to estimate when generic competition will enter.
Since patent entries in the Orange Book are not verified by FDA, applicant holders can, wittingly or not, add false information or non-applicable patents to the Orange Book. This can drastically extend the exclusivity period for a drug, delaying generic entry and keeping prices artificially high. The FTC has taken an increasingly active interest in challenging improper patent listings13 and has identified hundreds of potentially invalid listings in recent years.
Today, the Orange Book is available as an annual publication and a monthly Electronic Orange Book download. For analysts who need the data in structured, analysis-ready form, the RxDataLab FDA Exclusivity Dataset integrates the Electronic Orange Book with USPTO patent metadata, FDA drug label indication text, and a monthly change log tracking every patent modification, exclusivity grant, and application removal since October 2023. Full documentation is at rxdatalab.com/docs/fda-exclusivity/.
See 45 FR 72582 for initial rule establishing the orange book. ↩︎ ↩︎
The campaigns, including motion pictures narrated by popular actors are described in 44 FR 2932 p184. ↩︎
The FDA defines a therapeutic equivalents as one that contains the same active ingredients and method of action, and has undergone approval through an abbreviated new drug application (ANDA). ↩︎
For a more detailed history and the request for comments on the proposed rule, initially published on January 12, 1979, see 44 FR 2932 ↩︎ ↩︎
The 1979 FTC Staff report (PDF) titled “Drug Product Selection” contains a detailed history of antisubstitution laws, the transition to substitution laws, pricing, and estimated savings from adopting substitution laws. https://www.ftc.gov/reports/staff-report-drug-product-selection ↩︎
see New York Drug List, April 1, 1978 p185 44 FR 2932 ↩︎
The orange color for Halloween publication was described by Captain Kendra Stewart in a perspective piece looking back at 40 years of Orange Book publications ↩︎
Commonly known as the Hatch-Waxman Amendments to the FD&C act, this law formalized drug exclusivity, generic approval via ANDA’s (505j approval) and much more. ↩︎
See more in the Orange Book preface. ↩︎ ↩︎
Tentative approvals can be found in the Drugs@FDA database. ↩︎
See the FTC Patent Listing Challenges from April 2024 and Statement on Improper Patent Listings ↩︎ ↩︎
Get notified when we publish new research.
Primary-source analysis of biotech filings, trials, and fund positioning. No roundups, no filler.
Have a specific question about our methodology or data? Get in touch →